Wie genau läuft der Usability Engineering Prozess ab? Was fordert die IEC 62366-1 von Ihnen? Was genau müssen Sie nachweisen?
In diesem Artikel klären wir genau diese Fragen und schauen uns den Usability Engineering Prozess Schritt für Schritt an. Wir klären, was Sie als Hersteller von Medizinprodukten leisten müssen, um am Ende eine gute Usability und eine sichere Bedienung Ihres Produkts nachweisen zu können. Dabei schauen wir immer wieder in die IEC 62366-1 und zeigen Ihnen, welche Stellen Sie kennen und beachten müssen. Am Ende dieses Artikels haben Sie ein umfangreiches Verständnis von dem empfohlenen Usability Engineering Prozess und wissen, was von Ihnen verlangt wird und wie Sie das erfüllen (und nachweisen!).
Einleitung Teil 1 – Was ist eigentlich Usability Engineering?
Bevor wir nachfolgende tiefer in den geforderten Prozess einsteigen, möchten wir zunächst klären, was die IEC 62366-1 überhaupt unter Usability Engineering versteht. Dafür sehen wir uns die Definition aus dem Text an:
Usability Engineering ist die “Anwendung von Kenntnissen über menschliches Verhalten, Fähigkeiten, Einschränkungen und andere Merkmale in Bezug auf das Design von Medizinprodukten (Software eingeschlossen), Systemen und Aufgaben, um eine adäquate Usability zu erzielen“. (source: IEC 62366-1:2021-08)
Weiter wird die Anmerkung ergänzt: „Das Erreichen eines adäquaten Maßes an Usability kann zu einem akzeptablen Risiko bezogen auf die Benutzung des Medizinprodukts führen.“.
Usability Engineering für Medizinprodukte verfolgt also das Ziel, durch nutzerzentriertes Usability Design unakzeptable Risiken für Patienten, Nutzer und Dritte auszuschließen. Er will Use Errors herausfinden und diese dann minimieren.
Einleitung Teil 2 – Warum ist der Prozess so wichtig bei Medizinprodukten?
Für die Zulassung Ihres Medizinprodukts in Europa ist die Voraussetzung, dass Sie alle Anforderungen der MDR erfüllen. Diese beinhalten auch die grundlegenden Leistungs- und Sicherheitsanforderungen (Annex I), die Sie mithilfe der Einhaltung der IEC 62366-1 umsetzen können. Der Usability Engineering Prozess ist also ein sehr gutes Hilfsmittel dafür, einen wichtigen Teil der Anforderungen der MDR einzuhalten.
Welch große Rolle der Usability Engineering Prozess nach IEC 62366-1 für die Entwicklung von Medizinprodukten spielt, wird klar, wenn man sich vor Augen führt, dass die IEC 62366-1 von den Zulassungsstellen der unterschiedlichen Märkte anerkannt wird. Die FDA zum Beispiel führt die Norm als Recognized Consensus Standard auf.
Die starke normative Regulierung der Produktentwicklung ergibt Sinn, wenn man bedenkt, dass Fehlbedienungen von Medizinprodukten im schlimmsten Falle zum Tod führen können. Die falsche Interpretation von Daten, eine falsche oder zu späte Behandlung durch Bedienfehler – all das soll vermieden werden.
Um das Risiko des Produkts auch richtig einschätzen zu können, ist der Usability Engineering Prozess eng verbunden mit dem Risk Management. Dabei gilt: Die Usability Experten sind für die Bediensicherheit verantwortlich, das Risk Management für die Sicherheit des kompletten Produkts. Der Usability Engineering Prozess (wie alle anderen Prozesse) muss dabei durch nachgewiesen geeignetes Personal begleitet werden.
Tipp „von der Pflicht pragmatisch zur Kür“: Da Sie Usability Engineering inklusive des Nachweises der sicheren Bedienung (z.B. über formative und summative Nutzertests) betreiben müssen, lohnt es sich für Sie darüber hinaus auch über die Verbesserung der User Experience Ihres Medizinprodukts nachzudenken. Mit dem richtigen Ansatz kann diese direkt „mitgemacht“ werden. So nutzen Sie die Chance, die Ihnen der vorgeschriebene menschzentrierte Entwicklungsprozess bietet, um sich mit einer herausragenden UX gegenüber der Konkurrenz besser zu positionieren. Bestrebungen für eine bessere UX sind allerdings keine Vorschrift der IEC 62366-1.
Einleitung Teil 3 – Brauche ich überhaupt einen Usability Engineering Prozess?
Die schnelle Antwort: „ja“. Die etwas Ausführlichere: Die IEC 62366-1 (mit dem Titel: „Anwendung der Gebrauchstauglichkeit auf Medizinprodukte“) ist eine internationale Norm, die Usability Anforderungen für die Entwicklung von Medizinprodukten festlegt. Um sicherzustellen, dass diese Anforderungen auch eingehalten werden, wird hier der Usability Engineering Prozess gefordert. Sie brauchen einen Usability Engineering Prozess, der alle Anforderungen der IEC 62366-1 erfüllt, wenn Sie Konformität zur Norm erklären wollen. Es muss allerdings nicht genau der Prozess aus der Norm sein. Sie dürfen immer auch mehr machen. Allerdings müssen Sie die in der Norm geforderten Minimalkriterien erfüllen.
Einschub: Wer nicht dokumentiert, verliert!!!
Sie haben es bereits an den drei Ausrufungszeichen erkannt: Diese Info ist besonders wichtig! Um es ganz plump zu sagen: Keine (richtige) Dokumentation, keine Zulassung. Deswegen möchten wir hier, bevor wir uns den Prozess im Detail anschauen, darauf hinweisen, dass all Ihre Unternehmungen innerhalb des Usability Engineering Prozesses im sogenannten Usability Engineering File dokumentiert werden müssen.
Das Usability Engineering File (oder Gebrauchstauglichkeitsakte) wird von der IEC 62366-1 definiert als: „Sammlung von Aufzeichnungen und anderen Dokumenten, die während des Usability Engineering Prozesses erstellt werden“. (source: IEC 62366-1:2021-08, Kapitel 3.18)
Die Form ist dabei relativ frei: In Kapitel 4.2 wird ausgeführt, dass die Teile, die die Inhalte des Usability Engineering Files bilden, Teile anderer Dokumente und Akten sein können. Weitere Angaben zur Form werden nicht gemacht.
Der Prozess – Schritt für Schritt nach IEC 62366-1
Schauen wir uns doch einmal den Usability Engineering Prozess nach IEC 62366-1 im Überblick an. Die Grafik kann auf den ersten Blick etwas erschlagend wirken, ist aber bei näherer Betrachtung dann doch weit weniger einschüchternd als anfangs gedacht. Eines vorweg: Die Grafik zeigt sowohl den Prozess für das Usability Engineering (rechter Bereich), als auch den des Risk Managements (linker Bereich).
Wir werden diesen Prozess nun Schritt für Schritt „ablaufen“ und dabei immer wieder einen Blick in die Norm werfen.
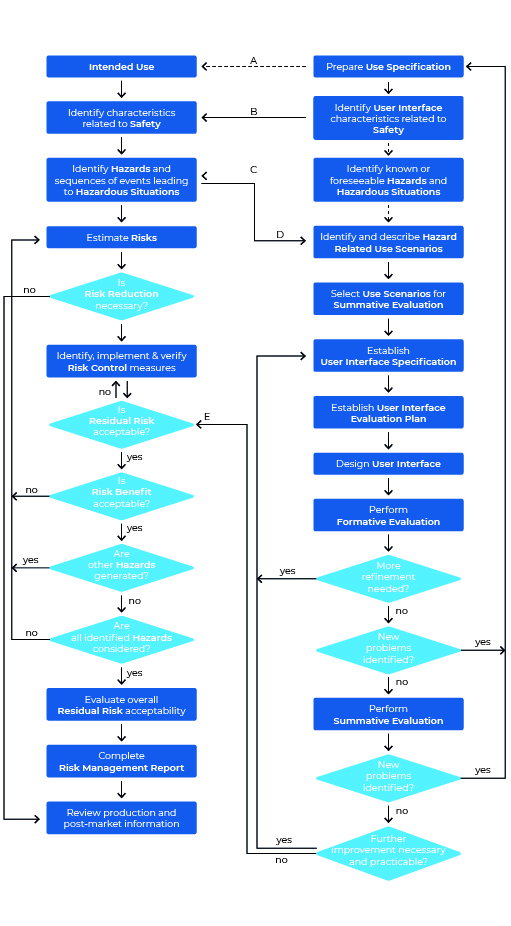
Schritt 1: Prepare Use Specification
Auch eine Usability-Reise beginnt mit dem ersten Schritt. Im Usability Engineering Prozess heißt dieser erste Schritt „Erstellen der Use Specification“. Direkt mit dem ersten Schritt müssen Sie die Dokumentation aller Maßnahmen beginnen, denn: „Die Einhaltung wird durch Einsichtnahme des Usability Engineering Files geprüft“ (source: IEC 62366-1:2021-08) – soll heißen: Sie müssen alle Ihre Schritte des Prozesses in einem Usability Engineering File festhalten (Sie erinnern sich: Wer nicht dokumentiert, verliert).
Woraus besteht die Use Specification? Die IEC 62366-1 fordert die Festlegung der folgenden Inhalte:
- Die vorgesehene medizinische Indikation. Im Englischen: Intended Medical Purpose. Hier legen Sie fest, wofür das Medizinprodukt verwendet werden soll. Der Intended Medical Purpose kann auch einen Ausschluss beinhalten (also die klare Information darüber, wofür das Medizinprodukt eben nicht verwendet werden darf!).
- Die vorgesehenen Nutzer. Im Englischen: Intended User Profile. Hier geht es darum, von wem das Produkt verwendet werden soll. Es reicht aber nicht aus, nur die Nutzergruppe zu benennen, Sie müssen diese auch noch in den relevanten Kriterien beschreiben.
- Die vorgesehenen Patientengruppen. Im Englischen: Intended Patient Population. An wem wird das Medizinprodukt verwendet? Die Norm liefert beispielhafte Abgrenzungskriterien für die Patientengruppen. Darunter Gesundheitszustand und Alter.
- Die vorgesehene Nutzungsumgebung. Im Englischen: Intended Use Environment. In welchem Nutzungskontext wird das Medizinprodukt verwendet werden? Auch die Intended Use Environment muss in den relevanten Kriterien beschrieben werden.
- Das vorgesehene Körperteil oder vorgesehene Gewebetyp, an dem das Produkt Anwendung finden soll. Im Englischen: Type of Tissue/ Part of Body. Hier geht es darum, festzuhalten, an welcher Körperstelle oder welcher Art von Gewebe das Medizinprodukt eingesetzt werden soll.
- Das Funktionsprinzip des Medizinprodukts. Im Englischen: Operating Principle. Wie funktioniert das Medizinprodukt und auf welche Art und Weise erbringt es seine Wirksamkeit?
(source: IEC 62366-1:2021-08; Kapitel 5.1)
Wie Sie diese Inhalte erarbeiten können, legt die Norm nicht fest. Wir empfehlen vor allem im Bereich der vorgesehenen Nutzer- und Patientengruppen sowie der Nutzungsumgebung eine gut dokumentierte Nutzungskontextanalyse.
Schritt 2: Identify User Interface Characteristics related to safety and potential Use Errors:
Kapitel 5.2. fordert von Ihnen, dass Sie als Teil der Risikoanalyse alle Teile und Eigenschaften des User Interfaces identifizieren, die mit der Sicherheit des Medizinprodukts zusammenhängen. (source: IEC 62366-1:2021-08; Kapitel 5.2).
Das heißt: Sie müssen im Zuge Ihres Usability Engineerings definieren und herausfinden, welche Eigenschaften der Nutzeroberfläche riskant für den Nutzer, für Patienten und Dritte sein können. Daraus leiten Sie dann Bedienfehler ab, die während der Nutzung entstehen können. Ein Beispiel: Sie entwerfen ein Interface, das kritische Messdaten ausspielt. Ein anzunehmender Bedienfehler wäre eine fehlerhafte oder ausbleibende Interpretation von angezeigten Daten aufgrund fehlender Vorkenntnisse einiger Nutzer.
Für die Risikoanalyse verweist die Norm auf die ISO 14971:2019 (Risk-Management). In dieser Norm finden Sie weitere Hinweise, wie Sie leichter die sicherheitsrelevanten Aspekte Ihres Interfaces identifizieren können.
Ein sehr brauchbares Tool zur Identifikation von Bedienfehlern ist die Task Analyse, sowie die Use Related Risk Analysis, deren Durchführung für den US-Amerikanischen Markt gefordert wird. Diese hilft Ihnen, die sicherheitskritischen Charakteristika Ihrer Benutzeroberfläche herauszufinden. Das Ziel der Taskanalyse ist es, die Sicherheitsrisiken der Bedienung herauszufinden. Die Sicherheitsrisiken des gesamten Produkts (also auch Faktoren wie mögliche Strahlung etc.) werden durch das Risk Management herausgefunden.
Schritt 3: Identify Known or forseeable Hazards and Hazardous Situations:
Kapitel 5.3 der Norm befasst sich mit dem „Ermitteln bekannter oder vorhersehbarer Gefährdungen und Gefährdungssituationen“. Auch hier sind Sie wieder für die bedienungsrelevanten Aspekte des Produkts verantwortlich (sprich, allem, was Usability-bezogen ist).
Bei der Ermittlung der Gefährdungen und Gefährdungssituationen verlangt die Norm von Ihnen, dass Sie die folgenden drei Dinge berücksichtigen:
- Die zuvor erarbeitete Use Specification Ihres Produkts.
- Die zuvor herausgefundenen Use Errors.
- Alle Informationen über Hazards und Hazardous Situations, die durch Medizinprodukte ähnlichen Typs bekannt sind. Hierzu können sowohl Wettbewerberprodukte als auch eigene Produkte einer älteren Generation zählen.
(source: IEC 62366-1:2021-08, Kapitel 5.3)
Dabei sind alle vorhersehbaren Gefährdungen und Gefährdungssituationen herauszufinden, die Nutzer, aber auch Patienten und Dritte betreffen können.
Schritt 4: Identify and describe Hazard-Related Use Scenarios
Aus den im Schritt zuvor identifizierten Gefährdungen und Gefahrensituationen werden jetzt die damit verbundenen gefährdungsbezogenen Nutzungsszenarien (Hazard-Related Use Scenarios) abgeleitet. Für Ihr Usability Engineering File müssen Sie diese ermitteln und beschreiben. Die Beschreibung muss laut Norm Folgendes enthalten:
- Alle Aufgaben des Szenarios.
- Die Reihenfolge der Aufgaben.
- Den Schweregrad des damit verbundenen Schadens.
(source: IEC 62366-1:2021-08, Kapitel 5.4)
Sie müssen also nicht nur die Hazard-Related Use Scenarios ermitteln, es muss auch eine Einschätzung des Schweregrads des möglichen Schadens erfolgen. Das Risk Management entscheidet dann, welche der Risiken akzeptabel sind und bei welchen Risiken Sie sich mit deren Reduktion und Beherrschung beschäftigen müssen (es werden also die Teile Ihres Produkts identifiziert, die überarbeitet werden müssen, um das Risiko bei der Nutzung zu beherrschen). Denken Sie daran, die MDR fordert von Ihnen, das Risiko so gering wie möglich zu halten, ohne das Risiko-Nutzen-Verhältnis negativ zu beeinflussen.
Schritt 5: Select Hazard-Related Use Scenarios for Summative Evaluation:
An den hier ausgewählten Szenarien wird in der summativen Evaluation eine abschließende Bewertung der Bediensicherheit Ihres Produkts vorgenommen. Das passiert, indem diese Szenarien in einer summativen Evaluation (z.B. einem Usability Test) überprüft werden.
Aber welche Szenarien müssen Sie auswählen? Entweder Sie testen alle gefährdungsbezogenen Szenarien oder aber eine begründete Auswahl, basierend auf dem Schweregrad der möglichen Schäden und „anderen Umständen, die spezifisch für das Medizinprodukt und den Hersteller sind“ (source: IEC 62366-1:2021-08, Kapitel 5.5).
Sie müssen laut Norm nicht nur die Auswahl der Hazard Related Use Scenarios für Ihre summative Evaluation im Usability Engineering File dokumentieren, sondern auch das gewählte Auswahlschema und eine ausreichende Begründung für die Wahl des jeweiligen Schemas.
Schritt 6: Establish User Interface Specification:
Das Erstellen der User Interface Specification muss laut Norm unter Berücksichtigung der Use Specification, der vorhersehbaren Use Errors und der gefährdungsbezogenen Use Scenarios stattfinden.
Die Norm fordert drei inhaltliche Punkte von Ihnen für die User Interface Specification:
- Eine Dokumentation aller testbaren technischen Anforderungen, die für die Nutzeroberfläche relevant sind.
- Es muss dokumentiert werden, ob Begleitdokumente für Ihr Medizinprodukt vorliegen. (Zum Beispiel Handbücher oder (Kurz-)Anleitungen).
- Es muss dokumentiert werden, ob ein Nutzertraining für Ihr Medizinprodukt notwendig ist.
(source: IEC 62366-1:2021-08, Kapitel 5.6)
Schritt 7: Establish User Interface Evaluation Plan:
Der User Interface Evaluation Plan ist ein Dokument, das festlegt, wie Sie prüfen werden, ob das Interface Ihres Medizinprodukts Risiken mit sich bringt. Sie bestimmen also, welche Teile des Interfaces wie getestet werden. Dabei müssen Sie die Methoden für alle formativen Evaluationen und der summativen Evaluation festhalten. Der User Interface Evaluation Plan muss Teil des Usability Engineering Files sein.
Entscheiden Sie sich für Usability Tests, verlangt die IEC 62366-1 eine ausführliche Dokumentation der Testbedingungen. Hierzu zählen die vorgesehenen Nutzergruppen (samt einer Begründung für die jeweilige Gruppierung), die Testumgebung (und andere Benutzungsbedingungen), mögliche Begleitdokumentation während des Tests und (falls nötig) die Maßnahmen zum Nutzertraining und deren Zeitabstand zwischen Training und Anwendung.
Die Norm lässt Ihnen offen, ob Sie in einer simulierten oder realen Nutzungsumgebung testen.
Viele Teile Ihres Interfaces müssen Sie mit Usability Tests (oder anderen geeigneten Methoden) evaluieren. Zum Beispiel müssen Sie hier die richtige Reaktion der Nutzer auf eine Fehlermeldung testen. Manche Teile allerdings können auch durch einen formalen Abgleich der Anforderungen mit dem Interface überprüft werden (z. B. eine bestimmte Größe eines Designelements). Dies kann durch eine Usability Verifizierung geschehen.
Schritt 8: Design User Interface:
Jetzt muss das User Interface Design entsprechend des gesammelten Wissens aller vorangegangenen Schritte umgesetzt werden. Vorgaben an das Design liefert die Norm nicht. Allerdings müssen zu diesem Zeitpunkt auch schon die Begleitdokumentation und die Nutzertrainings ausgestaltet werden.
Ziel ist es hier, alle aus den vorherigen Schritten abgeleiteten Gestaltungsempfehlungen in ein erstes Design umzusetzen, das Ihnen die formativen Evaluationen erlaubt, die bald folgen.
Schritt 9: Perform Formative Evaluations:
In diesem (nicht vorgeschriebenen) Schritt des Usability Engineering Prozesses nach IEC 62366-1 testen Sie Ihr Design formativ mit echten Nutzern. Dafür verwenden Sie einen Prototyp Ihres Produkts. Was genau geprüft werden soll, haben Sie bereits im User Interface Evaluation Plan festgelegt.
Ihr Ziel ist es jetzt, herauszufinden, ob Ihr Produkt (kritische) Bedienfehler generiert. Diese gilt es dann anschließend durch das Design zu minimieren oder auszuschließen (wo möglich). Sie prüfen also auch, ob Ihre Produktentwicklung auf dem richtigen Weg ist. Ihr Mindset: Sie wollen lernen und verstehen, welche Fehlbedienungen Ihr Produkt noch zutage fördert. Ziel ist es, Ihr Produkt besser und vor allem sicher zu machen.
Bei der Methodenwahl zur Umsetzung haben Sie freie Wahl. Es empfiehlt sich jedoch formativ mit Usability Tests zu testen, wenn die summative Evaluation ebenfalls ein Usability Test ist. So sind Sie vor unschönen Überraschungen während der abschließenden Prüfung sicher.
Wurden kritische Bedienfehler gefunden? Sind weitere Produktverbesserungen nötig? Haben Sie an diesem Punkt eine der beiden Fragen mit „ja“ beantwortet, gehen Sie im Usability Engineering Prozess soweit zurück wie notwendig und durchlaufen ihn ab diesem Punkt iterativ, bis keine neuen und kritischen Probleme bei der Nutzung mehr auftreten. Ist das der Fall, sind Sie bereit für den letzten großen Schritt: die Durchführung der summativen Evaluation.
Schritt 10: Perform summative Evaluation of the Usability of the User Interface:
Der letzte Schritt des Usability Engineering Prozesses ist die Durchführung der summativen Evaluation samt deren Nachbearbeitung. Für den abschließenden und bewertenden Test müssen Ihre Nutzer so geschult werden, wie Sie es auch nach Markteinführung würden. Ebenso muss Ihr Produkt fertig oder in Nullserie vorliegen, um die summative Evaluation durchzuführen.
Ziel ist es, nachzuweisen, dass das Medizinprodukt (und dessen Interface) ohne unakzeptable Restrisiken für Nutzer, Patienten und Dritte zu benutzen ist. Getestet werden die zuvor von Ihnen festgelegten gefährdungsbezogenen Nutzungsszenarien.
Zudem können Sie auch bereits bestehende Daten heranziehen: “Für die summative Evaluation kann der Hersteller Daten nutzen, die aus summativen Evaluationen von Produkten mit vergleichbarem User Interface gewonnen wurden, zusammen mit einer Begründung, wie diese Daten angewendet werden können.“ (Source: IEC 62366-1:2015, Kapitel 5.9)
Nach der Durchführung werden die Daten dahingehend ausgewertet, dass Sie alle aufgetretenen Bedienfehler und die daraus resultierenden (möglichen) Auswirkungen analysiert haben. Wenn durch einen der gemachten Bedienfehler eine Gefährdungssituation entstehen könnte, muss eine Ursachenanalyse durchgeführt werden. Dazu nutzen Sie die aus den Beobachtungen und Nachbefragungen der Studienteilnehmer gewonnenen Daten.
Sind neue Bedienfehler, Gefährdungen oder Gefährdungssituationen aufgetreten, müssen Sie abwägen, ob diese eine Überarbeitung des Produkts notwendig machen oder nicht. Hier noch einmal die dringende Empfehlung, alle gefährdungsbezogenen Nutzungsszenarien in formativen Tests ausgiebig zu testen, um sicherzugehen, dass in der summativen Evaluation keine unschönen und kostspieligen Überraschungen entstehen.
Fällt Ihr Produkt durch, wird die summative Evaluation zur formativen und der Usability Engineering Prozess geht weiter, bis Ihr Produkt sicher genug für die summative Studie ist.
Die Norm verpflichtet Sie dann dazu, zu dokumentieren, „warum eine Verbesserung nicht notwendig oder nicht praktikabel ist“ (source: IEC 62366-1:2021-08, Kapitel 5.9).
Wurde anhand der Daten das Restrisiko bestimmt, haben Sie das Ende des Usability Engineering Prozesses erreicht.
Weitere Tipps für die Durchführung der summativen Evaluation nach IEC 62366-1 finden Sie in unserem Artikel „17 Dos & Dont’s bei der Durchführung von summativen Usability Evaluationen – Wie Sie Ihre Ergebnisse direkt verbessern, sich die Zulassung sichern und die IEC 62366-1 einhalten“.
Usability Engineering nach anderen Normen und Gesetzen:
Die amerikanische Food and Drug Administration (FDA) ist sogar noch etwas direkter als MDR und IVDR: Sie fordert ganz klar den Usability Engineering Prozess (bzw. Human Factors Engineering Prozess) und liefert dafür sogar ein umfangreiches Guidance Dokument.
Diese werden wir allerdings in gesonderten Artikeln genauer unter die Lupe nehmen.
Fazit
Der Usability Engineering Prozess ist in der IEC 62366-1 sehr ins Detail beschrieben und immer eng mit dem Risk Management verbunden. Wichtig sind die richtige Umsetzung und die lückenlose Dokumentation! Alles ist auf die Einschätzung und Kontrolle der Bedienrisiken ausgerichtet.
Haben Sie den Prozess gewissenhaft durchlaufen und alles richtig dokumentiert, gibt es im Bereich Usability keine Stolpersteine mehr für Ihr Medizinprodukt.
Für welches Medizinprodukt müssen Sie Usability Engineering anwenden? Kommentieren Sie diesen Beitrag oder melden Sie sich über unser Kontaktformular.


