How exactly does the Usability Engineering process work? What does IEC 62366-1 require of you? What exactly do you have to prove?
In this article, we clarify exactly these questions and take a step-by-step look at the Usability Engineering process. We clarify what you, as a manufacturer of medical devices, have to achieve in order to be able to prove good Usability. In doing so, we look again and again at IEC 62366-1 and show you which parts you need to know and observe. By the end of this article, you will have a comprehensive understanding of the recommended Usability Engineering process and know what is required of you and how to meet (and prove!) it.
Introduction Part 1 – What is Usability Engineering?
Before we dig deeper into the required process, we would like to clarify what IEC 62366-1 actually defines as Usability Engineering. For this we look at the definition from the text:
Usability Engineering is the ” application of knowledge about human behaviour, abilities, limitations, and other characteristics to the design of MEDICAL DEVICES (including software), systems and TASKS to achieve adequate USABILITY.” (source: IEC 62366-1:2021-08)
Further, the note adds, “Achieving adequate USABILITY can result in acceptable RISK related to use”.
Usability Engineering for medical devices thus pursues the goal of eliminating unacceptable risks to patients, users, and third parties through user-centered Usability Design. It aims to find out Use Errors and then minimize them.
Introduction Part 2 – Why is the process so important for medical devices?
To get your medical devices regulatory approval in Europe, it is a prerequisite that you meet all the requirements of the MDR. These include the general performance and safety requirements (Annex I), which you can implement by complying with IEC 62366-1. So the Usability Engineering process is a very good tool to comply with an important part of the MDR requirements.
The importance of the IEC 62366-1 Usability Engineering Process for the development of medical devices becomes clear when you realize that IEC 62366-1 is recognized by the regulatory bodies of the different markets. The FDA, for example, lists the standard as a Recognized Consensus Standard.
The strong normative regulation of product development makes sense when one considers that misuse of medical devices can lead to death in the worst case scenario. The wrong interpretation of data, incorrect or too late treatment due to operating errors – all this should be avoided!
In order to be able to assess the risk of the product correctly, the Usability Engineering process is closely linked to Risk Management. The Usability experts are responsible for the safety of use, while the Risk Management is responsible for the safety of the complete product. The Usability Engineering process (like all other processes) must be accompanied by certified personnel.
Tip: Since you have to perform Usability Engineering including the proof of safe use (e.g. via formative and summative user tests), it is also worthwhile for you to think about improving the User Experience of your medical product. With the right approach, this can be done simultaneously. This way, you can take advantage of the opportunity that the mandatory human-centered development process offers you to better position yourself against the competition with an outstanding UX. However, aspirations for a better UX are not a requirement of IEC 62366-1.
Introduction Part 3 – Do I even need a Usability Engineering process?
The quick answer: “yes”. The more detailed one: IEC 62366-1 (titled: “Application of Usability to Medical Devices”) is an international standard that specifies Usability requirements for the development of medical devices. To ensure that these requirements are met, the Usability Engineering process is required here. You need a Usability Engineering process that meets all the requirements of IEC 62366-1 if you want to declare compliance with the standard. However, it does not have to be exactly the process from the standard. You are always allowed to do more. However, you must meet the minimum criteria required by the standard.
Add-in: Without the right documentation, you fail!!!
You have already recognized it by the three exclamation marks: This info is especially important! To put it bluntly: No (correct) documentation, no clearance of your product. Therefore, before we look at the process in detail, we would like to point out that all your undertakings within the Usability Engineering process must be documented in the so-called Usability Engineering File.
The Usability Engineering File is defined by IEC 62366-1 as: ” set of RECORDS and other documents that are produced by the USABILITY ENGINEERING PROCESS “. (source: IEC 62366-1:2021-08, chapter 3.18)
The form is relatively free: Chapter 4.2 states that the parts forming the contents of the Usability Engineering File may be parts of other documents and files. No further information about the form is given.
The process – step by step according to IEC 62366-1
Let’s take a look at the Usability Engineering process according to IEC 62366-1. The graphic may seem a bit overwhelming at first glance, but on closer inspection it is far less intimidating than initially thought. One thing first: The graphic shows both the process for Usability Engineering (right area) and that of Risk Management (left area).
We will now ” walk through” this process step by step, taking a look at the standard every now and then.
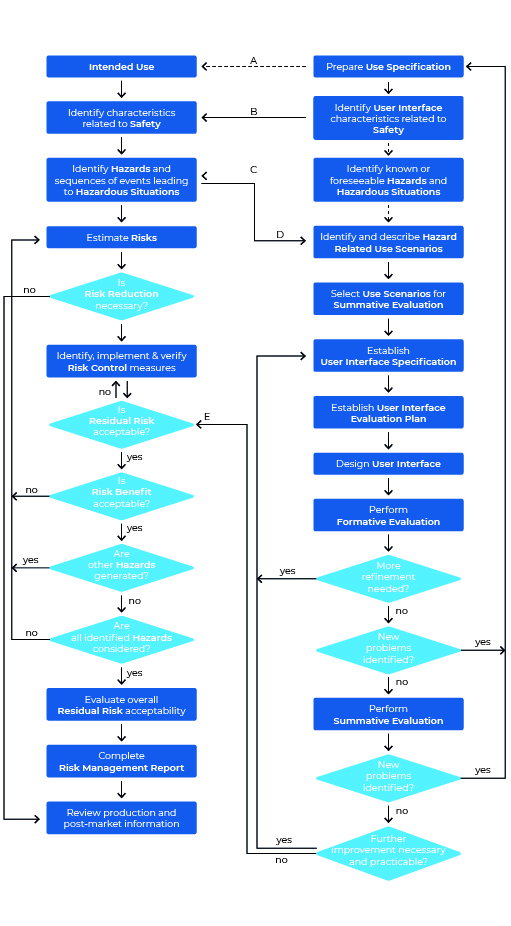
Step 1: Prepare Use Specification
Even a Usability journey begins with the first step. In the Usability Engineering process this first step is called “Prepare Use Specification”. Directly with the first step you have to start the documentation of all actions, because the compliance is checked by inspection of the Usability Engineering File (source: IEC 62366-1:2021-08) – this means: You have to record all your steps of the process in a Usability Engineering File (you remember: If you don’t document, you fail).
What does the Use Specification consist of? IEC 62366-1 requires the following content to be specified:
- The Intended Medical Purpose. This is where you specify what the medical device is to be used for. The intended medical purpose can also include an exclusion (i.e. clear information about what the medical device must not be used for).
- Intended User Profile. This is about who the product is intended to be used by. However, it is not enough just to name the user group, you must also describe it in terms of the relevant criteria.
- The Intended Patient Population. On whom will the medical device be used? The standard provides exemplary delimitation criteria for the patient groups. These include health status and age.
- The Intended Use Environment. In which context will the medical device be used? The Intended Use Environment must also be described in the relevant criteria.
- The Type of Tissue/ Part of Body. The purpose here is to specify the body part or type of tissue on which the medical device is intended to be used.
- The operating principle of the medical device. How does the medical device work and how does it perform?
(source: IEC 62366-1:2021-08; chapter 5.1)
The standard does not specify how you can develop this content. We recommend a well-documented context of use analysis, especially in the area of the intended user and patient groups and the environment of use.
Step 2: Identify User Interface Characteristics related to safety and potential Use Errors:
Chapter 5.2. requires you to identify, as part of the risk analysis, all parts and characteristics of the User Interface that are related to the safety of the medical device. (source: IEC 62366-1:2021-08; chapter 5.2).
This means: In the course of your Usability Engineering you have to define and find out which properties of the User Interface can be hazardous for the user, for patients and for third parties. From this, you then derive use errors that can occur during use. An example: You design an Interface that displays critical measurement data. An assumed operating error would be an incorrect or omitted interpretation of displayed data due to a lack of prior knowledge on the part of some users.
For risk analysis, the standard refers to IEC 14971:2019 (Risk management). In this standard, you will find further guidance on how to more easily identify the security-related aspects of your Interface.
A very useful tool for identifying use errors is the Task Analysis, as well as the Use Related Risk Analysis, which is required to be performed for the US market. This helps you to find out the safety-related characteristics of your User Interface. The goal of the Task Analysis is to find out the security risks of the usage. The safety risks of the entire product (i.e. also factors such as possible radiation, etc.) are found out by Risk Management.
Step 3: Identify Known or forseeable Hazards and Hazardous Situations:
Chapter 5.3 of the standard deals with “Identifying known or foreseeable hazards and hazardous situations”. Once again, you are responsible for the aspects of the product that are relevant to use ( i.e., everything Usability-related).
In determining the hazards and hazardous situations, the standard requires you to consider the following three things:
– The previously developed Use Specification of your product.
– The previously identified Use Errors.
– All information about Hazards and Hazardous Situations known from medical devices of similar type. This can include competitor products as well as own products of an older generation.
(source: IEC 62366-1:2021-08, chapter 5.3)
All foreseeable hazards and hazardous situations that may affect users, but also patients and third parties, must be identified.
Step 4: Identify and describe Hazard-Related Use Scenarios
The associated hazard-related use scenarios are now derived from the hazards and hazardous situations identified in the previous step. For your Usability Engineering File, you must identify and describe these. According to the standard, the description must include the following:
– All tasks in the scenario.
– The sequence of the tasks.
– The severity of the associated harm.
(source: IEC 62366-1:2021-08, chapter 5.4)
So not only do you need to identify the Hazard-Related Use Scenarios, there also needs to be an assessment of the severity of the potential harm. Risk Management then decides which of the risks are acceptable and for which risks you need to address their reduction and control (i.e., it identifies the parts of your product that need to be revised to control the risk during use). Remember, the MDR requires you to keep the risk as low as possible without negatively impacting the risk-benefit ratio.
Step 5: Select Hazard-Related Use Scenarios for Summative Evaluation:
The scenarios selected here are used in the Summative Evaluation to make a final assessment of the Usability of your product. This is done by checking these scenarios in a Summative Evaluation (e.g. a Usability Test).
But which scenarios do you have to choose? Either you test all hazard-related scenarios, or you test a reasoned selection based on the severity of potential harm and other circumstances specific to the medical device and the manufacturer (source: IEC 62366-1:2021-08, Chapter 5.5).
According to the standard, you must document not only the selection of Hazard Related Use Scenarios for your Summative Evaluation in the Usability Engineering File, but also the chosen selection scheme and sufficient justification for the choice of the respective scheme.
Step 6: Establish User Interface Specification:
According to the standard, the User Interface Specification must be created, taking into account the Use Specification, the foreseeable Use Errors and the hazard-related Use Scenarios.
The standard requires three content points from you for the User Interface Specification:
- documentation of all testable technical requirements relevant to the user interface.
- documentation of whether there are any accompanying documents for your medical device. (For example, instructions for use, manuals or (brief) instructions).
- it must be documented whether user training is necessary for your medical device.
(source: IEC 62366-1:2021-08, chapter 5.6)
Step 7: Establish User Interface Evaluation Plan:
The User Interface Evaluation Plan is a document that defines how you will test whether the Interface of your medical device poses hazards. In other words, you determine which parts of the Interface will be tested and how. You must record the methods for all formative evaluations and the summative evaluation. The User Interface Evaluation Plan must be part of the Usability Engineering File.
If you decide to use Usability tests, IEC 62366-1 requires detailed documentation of the test conditions. This includes the intended user groups (including a justification for each grouping), the test environment (and other usage conditions), possible supporting documentation during the test, and (if necessary) the user training measures and their time interval between training and application.
The standard leaves it open to you whether you test in a simulated or real use environment.
You need to evaluate many parts of your interface with Usability tests (or other appropriate methods). For example, you may need to test the correct user response to an error message. Some parts, however, can also be verified by formally matching the requirements to the Interface (e.g., a specific size of a design element). This can be done by a Usability verification.
Step 8: Design User Interface:
Now the User Interface Design must be implemented according to the accumulated knowledge of all previous steps. The standard does not provide any specifications for the design. However, the accompanying documentation and user training must already be designed at this stage.
The goal here is to implement all the design recommendations derived from the previous steps into an initial design that will allow you to conduct the formative evaluations that will soon follow.
Step 9: Perform Formative Evaluations:
In this (non-mandatory) step of the Usability Engineering process according to IEC 62366-1, you formatively test your design with real users. You use a prototype of your product for this purpose. You have already defined what exactly is to be tested in the User Interface Evaluation Plan.
Your goal now is to find out whether your product generates (critical) use errors. These must then be minimized or eliminated (where possible) through the design. So, you also check if your product development is on the right track. Your Mindset: You want to learn and understand which use errors your product still brings to light. The goal is to make your product better and, above all, safer.
You are free to choose the methods for implementation. However, it is recommended to test formatively with usability tests, if the summative evaluation is also a usability test. This way you are safe from unpleasant surprises during the final test.
Were critical use errors found? Are further product improvements necessary? If you have answered “yes” to either of these questions at this point, go back as far as necessary in the Usability Engineering process and iterate through it from this point on until no new and critical problems occur during use. If this is the case, you are ready for the last big step: performing the summative evaluation.
Step 10: Perform summative Evaluation of the Usability of the User Interface:
The last step of the Usability Engineering process is the execution of the summative evaluation including its post-processing. For the final and evaluative test, your users must be trained as they would be after market launch. Likewise, your product must be ready or in pilot production to perform the summative evaluation.
The goal is to prove that the medical device (and its Interface) can be used without unacceptable residual risks for users, patients and third parties. The hazard-related use scenarios previously defined by you are tested.
In addition, you can also use existing data: For the Summative Evaluation, the manufacturer may use data obtained from summative evaluations of devices with comparable user interface, together with a justification of how these data can be applied. (Source: IEC 62366-1:2015, Chapter 5.9)
After implementation, the data is evaluated to the effect that you have analyzed all the use errors that occurred and the resulting (possible) effects. If a hazardous situation could arise as a result of one of the use errors made, a root cause analysis must be performed. To do this, use the data obtained from the observations and follow-up interviews of the study participants.
If new use errors, hazards or hazardous situations have occurred, you must weigh up whether or not these make it necessary to revise the product. Here again is the urgent recommendation to extensively test all hazard-related use scenarios in formative tests to ensure that no unpleasant and costly surprises arise in the summative evaluation.
If your product fails, the summative evaluation becomes formative and the Usability Engineering process continues until your product is safe enough for the summative study.
The standard then requires you to document why improvement is not necessary or not practical. (source: IEC 62366-1:2021-08, Chapter 5.9)
If the residual risk has been determined based on the data, you have reached the end of the Usability Engineering process.
For more tips on conducting summative evaluation according to IEC 62366-1, see our article “17 Dos & Dont’s for the execution of the summative Usability evaluation“.
Usability Engineering according to other standards and laws:
The American Food and Drug Administration (FDA) is even more direct than MDR and IVDR: It clearly requires the Usability Engineering process (or Human Factors Engineering process) and even provides a comprehensive guidance document for this.
However, we will take a closer look at this in separate articles.
Conclusion
The Usability Engineering process is described in great detail in IEC 62366-1 and is always closely linked to Risk Management. Correct implementation and complete documentation are important! Everything is geared towards the assessment and control of use hazards.
If you have gone through the process conscientiously and documented everything correctly, there will be no more stumbling blocks for your medical device in the area of Usability.
For which medical device do you need to apply Usability Engineering? Comment on this post or get in touch via our contact form.
You are welcome to discuss your specific case with us in a free strategy meeting. We look forward to hearing from you!


